
Científicos trazan las primeras rutas de expansión del covid-19 por el mundo
especiales

Un grupo de investigadores del Reino Unido y Alemania recurrió a técnicas de redes genéticas para reconstruir los primeros caminos evolutivos de covid-19 en humanos, así como para mapear parte de la propagación original del coronavirus desde Wuhan a Europa y América del Norte.
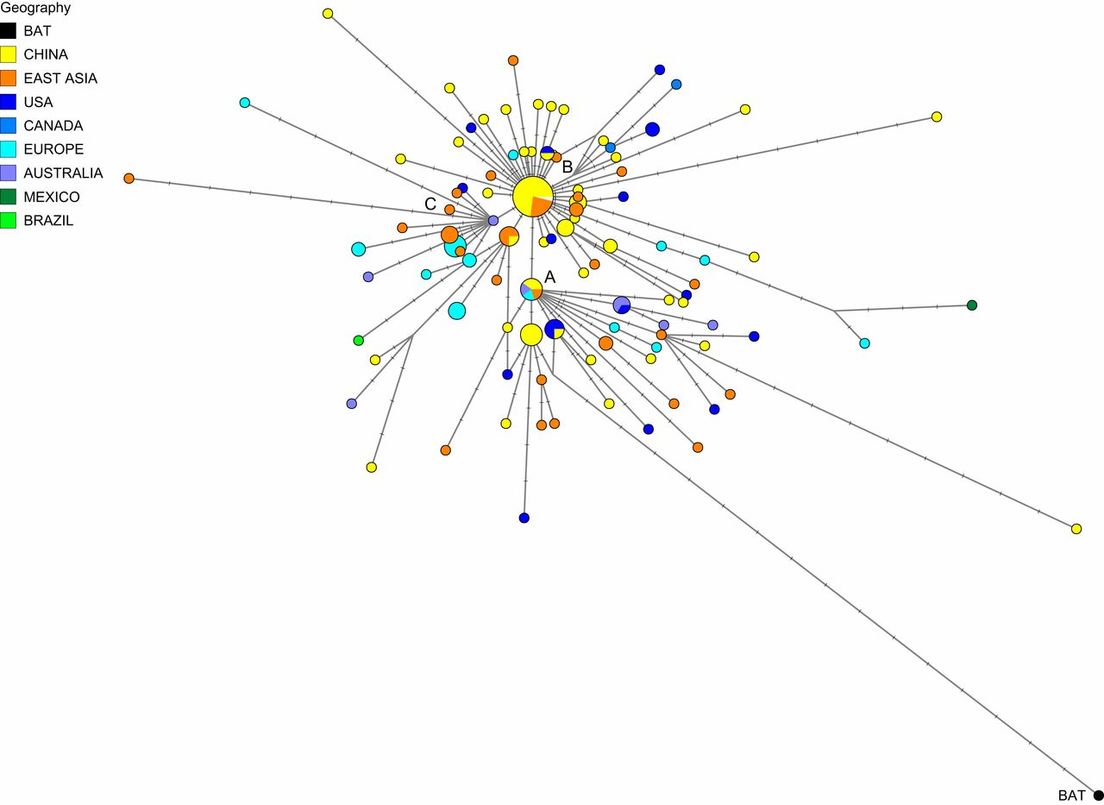
Al analizar los primeros 160 genomas completos del virus que se secuenciaron a partir de pacientes humanos, los científicos encontraron tres principales variantes del SARS-CoV-2, que denominaron 'A', 'B' y 'C'. La variante 'A', la más estrechamente relacionada con el virus encontrado en murciélagos y en pangolines, es descrita por los investigadores como "la raíz del brote", mientras que el tipo 'B' se deriva de 'A', separado por dos mutaciones, y la variante 'C' es una "hija" de 'B'.
Los hallazgos, publicados en la revista Proceedings of the National Academy of Sciences (PNAS), apuntan que la variante más cercana a la aislada en los murciélagos, la 'A', se encontró principalmente en pacientes de EE.UU. y Australia, pero, sorprendentemente, no en Wuhan, donde se originó el coronavirus. Aunque este primer tipo también estaba presente en esa ciudad china, la principal variante de virus en Wuhan fue la 'B', que también prevaleció en pacientes de todo el este de Asia, si bien no viajó mucho más allá de la región sin más mutaciones.
Entretanto, la variante 'C' aparece como el principal tipo europeo, que se encuentra en los primeros pacientes de Francia, Italia, Suecia y el Reino Unido. El análisis también sugiere que una de las primeras introducciones del virus en Italia se produjo a través de la primera infección alemana, documentada el 27 de enero, mientras que otra de las rutas tempranas de infección en el país transalpino estaba relacionada con un "grupo de Singapur".
Predecir futuros 'puntos calientes'
Los científicos argumentan que sus métodos "filogenéticos" podrían aplicarse a la última secuenciación del genoma del coronavirus para ayudar a predecir futuros puntos críticos mundiales de transmisión y auge de la enfermedad.
"El análisis de la red filogenética tiene el potencial de ayudar a identificar fuentes no documentadas de infección por covid-19, que luego se pueden poner en cuarentena para contener una mayor propagación de la enfermedad en todo el mundo", sostiene Peter Forster, genetista de la Universidad de Cambridge (Reino Unido) y autor principal del estudio.
Los casos confirmados de covid-19 en todo el mundo se acercan a los 2 millones, según los últimos datos actualizados por el Centro de Ciencia e Ingeniería de Sistemas de la Universidad Johns Hopkins, que monitorea las estadísticas a escala internacional.
El número de fallecidos por esa enfermedad ha superado 120.000, mientras que 464.398 afectados se han recuperado.
Destacado

Padrino López reafirmó que la FANB se mantiene en alerta y despliegue activo en todo el territorio para garantizar la soberanía y la integridad territorial ante cualquier intento de violación del espacio aéreo o marítimo. Foto: teleSUR.
EL TEMA
")









, Primer Secretario del Comité Central de Partido Comunista de Cuba y Presidente de la Republica, asiste al concierto del cantautor y fundador del Movimiento de la Nueva Trova, Silvio Rodríguez, en la escalinata de la Universidad de La Habana, el 19 de septiembre de 2025, como parte del inicio de una gira que lo llevara a cinco países de Latinoamérica. ACN FOTO/Luis JIMÉNEZ ECHEVARRÍA/sd")
Añadir nuevo comentario